Mecanismos de acción y resistencia a glucocorticoides en asma y enfermedad pulmonar obstructiva crónica
(Glucocorticoid action and resistance mechanisms in asthma and chronic obstructive pulmonary disease)
Alcibey Alvarado-González1, Isabel Arce-Jiménez2
Afiliación de los autores:
1Servicio de Neumología. Hospital San Juan de Dios, Caja Costarricense de Seguro Social. 2Estudiante Escuela de Medicina. Universidad de Costa Rica, San José, Costa Rica [email protected]
Fuentes de apoyo económico:
ninguna.

Glucocorticoides en asma y EPOC/ Alvarado-González y Arce-Jiménez
who smoke, as well as in all patients with chronic obstructive pulmonary disease. Several molecular mechanisms of glucocorticoid resistance have been identified. Alternative anti-inflammatory treatments that may allow for the control of these patients’ symptoms and drugs that may reverse molecular mechanisms of resistance are being investigated. Unfortunately, these therapies might be too specific to be effective, as is the case of dissociated glucocorticoids, where it has been difficult to dissociate anti-inflammatory effects from adverse effects.
Keywords: Asthma, chronic obstructive pulmonary disease, drug resistance, glucocorticoids,
inflammation.
Recibido: 08 de enero de 2013
El uso más uso común de los glucocorticoides (GC) en enfermedades respiratorias es en el tratamiento del asma.1 Los GC inhalados se han establecido como el tratamiento de primera línea en adultos y niños con asma persistente: la enfermedad inflamatoria crónica más común.2 Sin embargo, existe una minoría de pacientes con esta enfermedad que tienen poca o nula respuesta a los GC, aún en dosis altas. Esto también ocurre con la enfermedad pulmonar obstructiva crónica (EPOC).3
La presente revisión describe los mecanismos moleculares antinflamatorios de los GC, particularmente aquellos enfocados en el asma. También se discuten las bases moleculares de la resistencia a GC en asma, EPOC, sus implicaciones y alternativas terapéuticas.
Inflamación en asma: bases moleculares
Los pacientes con asma tienen un patrón específico de inflamación en las vías aéreas, caracterizado por la presencia de mastocitos degranulados, eosinófilos hipodensos y linfocitos Th2.4 Estas alteraciones son responsables de las manifestaciones clínicas de la entidad, que incluyen sibilancias intermitentes, disnea, tos y opresión torácica.5 Los GC controlan y previenen estos síntomas en muchos pacientes.
En el asma se pueden liberar aproximadamente 100 mediadores inflamatorios, dentro de los que se encuentran mediadores lipídicos, péptidos inflamatorios, citoquinas, quimosinas, enzimas inflamatorias y factores de crecimiento.6 La evidencia sugiere que las células estructurales de la vía aérea (p.ej.: células epiteliales, músculo liso, células endoteliales y fibroblastos) son la mayor fuente de estos mediadores.2
La expresión de mediadores inflamatorios es regulada por transcripción genética, la cual, a su vez, es controlada por factores de transcripción proinflamatorios, como el NF-kβ (factor nuclear kappa-beta), AP-1 (proteína activadora-1), NF-AT (factor nuclear de células T activadas) y STAT (transductores de señales y activadores de transcripción).7,8 Por ejemplo, el NF-kβ está marcadamente activo en las células epiteliales de pacientes asmáticos.5,9 Algunos de estos factores pueden ser activados por infección con rinovirus y por alérgenos, potenciales intensificadores de la inflamación asmática.10
Cuando los factores de transcripción proinflamatorios son activados, se unen a secuencias específicas de reconocimiento en el ADN. En esta interacción participan moléculas coactivadoras
Aceptado: 04 de julio de 2013
como: p300/CREB, CBP (proteína de unión a CREB), pCAF (factor activador de p300/CBP) y swi/snf (SwItch/sucrosa no fermentable). Estos complejos moleculares tienen actividad histona acetiltransferasa intrínseca (HAT), que conlleva a la expresión de los genes inflamatorios correspondientes (explicado en detalle adelante).11,12
Remodelación de la cromatina
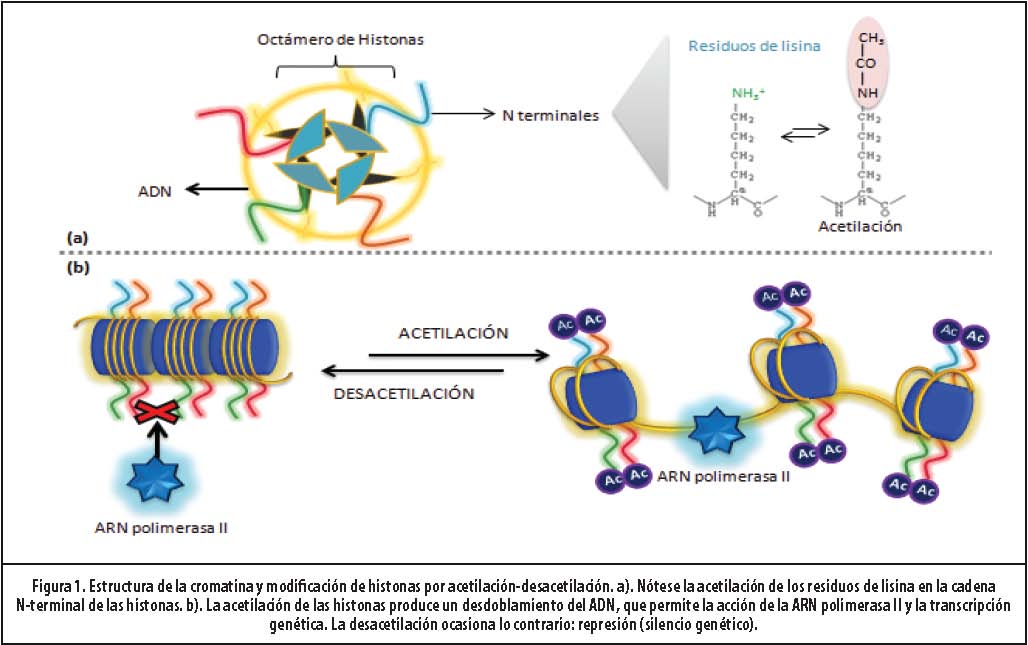
La estructura que presente la cromatina es crítica para la expresión y la represión de los genes. Estructuralmente, la cromatina está construida por unidades denominadas nucleosomas. A su vez, estos están constituidos por ADN asociado a un octámero de histonas (2H2A, 2H2B, 2H3, 2H4) y otras proteínas.13 (Figura 1)
En la célula en reposo, el ADN está ovillado alrededor del núcleo básico de histonas. Esta conformación obstaculiza los sitios de unión de la ARN polimerasa II, enzima encargada de la formación de ARN mensajero (p.ej., transcripción genética). Dicho entrelazamiento está dictado por atracción de cargas eléctricas. El ADN típicamente presenta una carga negativa, mientras el núcleo de histonas, una carga positiva. Es así como la cromatina adopta una distribución “cerrada”, que se asocia con la supresión de la expresión genética.10
Cada núcleo de histonas tiene una cadena N-terminal rica en residuos de lisina, que pueden ser acetilados y adquirir una carga eléctrica más negativa. Es así como la cromatina puede cambiar a la conformación “abierta”, perdiendo el enlace ADN-núcleo de histonas, dado que existe ahora una repulsión de cargas.14 Las hélices de ADN se desovillan y es posible la unión de la ARN polimerasa II a los sitios correspondientes en el ADN, formando ARN mensajero que viajará al citoplasma y emprenderá la síntesis de los mediadores inflamatorios o antinflamatorios, según el gen involucrado.15 Esta acetilación puede ser reversada por la actividad de las enzimas histonadesacetilasas (HDACs). Es así que resulta el papel de las HAT como coactivador y el de las HDAC como correpresor.16 (Figura 1) En biopsias de pacientes con asma, la actividad HAT está incrementada y la HDAC, reducida, lo que favorece la expresión de genes inflamatorios.17,18
El núcleo de histonas también pueden ser modificado por metilación (y desmetilación), fosforilación, nitración (estrés nitrativo), sumoilación y ubiquitinación.15,19 El concepto de “código de histonas” se refiere a estas modificaciones, que al alterar la estructura de la cromatina, se asocian a expresión o represión de genes.20-22
Efectos bioquímicos de los GC
Para ejercer su efecto, el GC debe difundir a través de la membrana plasmática y unirse a su receptor (RG) a nivel citoplasmático. Una vez activado el complejo (el GC unido al receptor), distintos mediadores facilitan su translocación al núcleo, donde se generan efectos directos e indirectos sobre el control de la inflamación.4 (Figura 2) A continuación se detalla este proceso.
1. Receptor de glucocorticoides (RG): el RG es codificado por un único gen, pero se reconocen varias isoformas creadas a través del empalme alternativo. En el citoplasma se encuentra el RGα, única isoforma del receptor que se une a los GC. La isoforma β se encuentra unida al ADN, y se desconoce su significancia funcional.1,23
Mientras no se encuentra estimulado por su ligando (GC), el RGα se mantiene unido a moléculas de tipo chaperonas (como las proteínas de choque de calor 70 y 90).4 La unión del ligando activa al RGα, el cual sufre un cambio estructural que libera las chaperonas.23 Con esto, se exponen sitios del receptor que señalan su translocación nuclear. Las proteínas de tipo importinas nucleares (p.ej., importina α e importina-13), permiten una rápida movilización del complejo.1
Para efectos didácticos, los mecanismos antinflamatorios de los GC se pueden estudiar desde 3 perspectivas: activación de genes antinflamatorios, desactivación de genes inflamatorios activados y efectos postranscripcionales. Los primeros dos modelos dependen, sobre todo, de la dosis de GC, y están íntimamente relacionados con la remodelación de la cromatina.
2. Activación genética: GC a dosis altas: en el núcleo, dos complejos GC-RGα homodimerizan y adquieren la capacidad de unirse a secuencias específicas de ADN. Los genes sensibles a GC tienen en su promotor secuencias de nucleótidos denominadas “elementos de respuesta a glucocorticoides” (GRE).24 La unión del homodímero al GRE permite modular la expresión genética: estimular (menos común, inhibir) la transcripción del gen específico. Esta activación es mediada por la interacción RG-ADN-moléculas transcripcionales coactivadoras (p.ej., CBP, SRC-2, pCAF). (Figura 2) Estas últimas poseen actividad acetiltransferasa intrínseca, lo que causa la acetilación de las histonas.23 (Figura 1)
Los genes que son activados por los GC incluyen los que codifican para el receptor β2 adrenérgico, inhibidor de la leucoproteasa secretora y MPK-1 (una fosfatasa-1 inhibidora de la MAPK).4 De esta forma, se producen proteínas antinflamatorias. Como se mencionó, la inhibición de la expresión genética es menos frecuente y está más relacionada con los efectos adversos de los GC.
3. Desactivación de genes inflamatorios activados: GCs a bajas dosis: en el control de la inflamación, la principal acción de los GC es la inhibición de la síntesis de mediadores inflamatorios.4,23,24 Dos mecanismos reprimen la expresión genética e interactúan con moléculas correpresoras, de
Glucocorticoides en asma y EPOC/ Alvarado-González y Arce-Jiménez
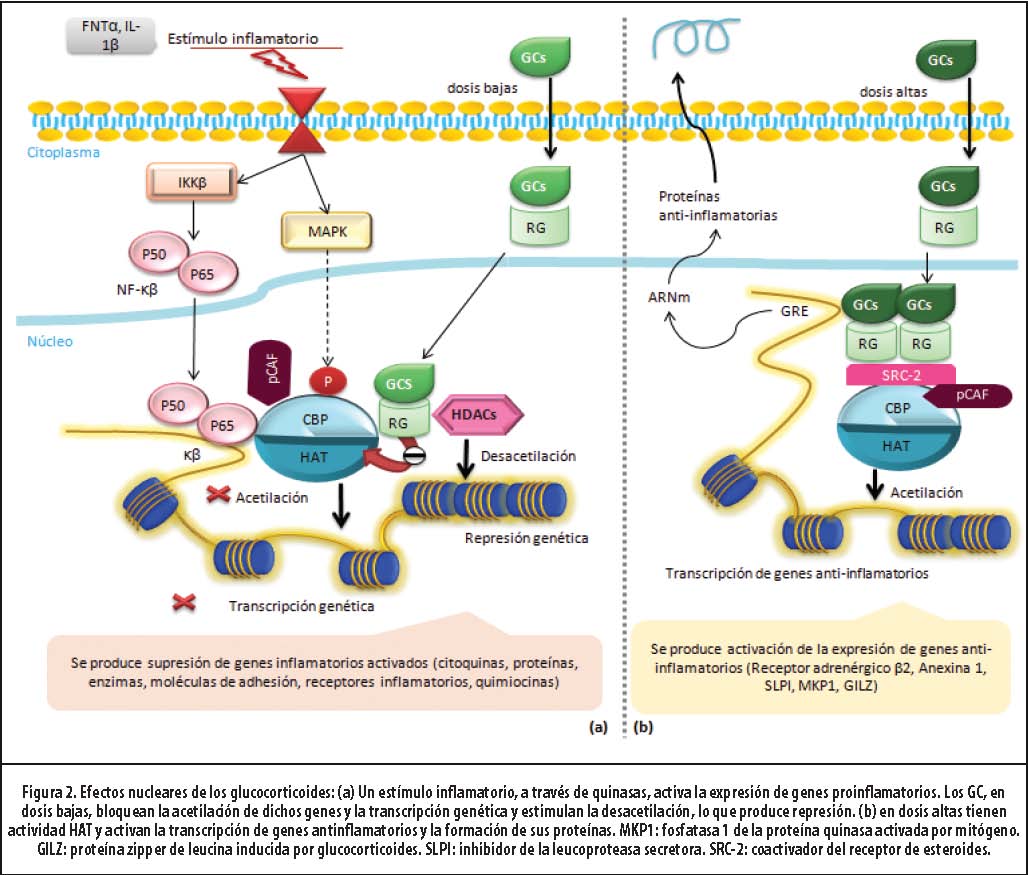
modo que contrarreste la respuesta coactivadora de factores proinflamatorios. Se limita así la acetilación de las histonas, la remodelación de la cromatina y la unión de la ARN polimerasa II.23 El mecanismo más importante se produce mediante el reclutamiento de la HDAC-2 y resulta de esta vía la supresión de los genes inflamatorios ya activados.23 ,24
Resistencia a los GC
El asma bronquial generalmente se controla con dosis bajas de GC. No obstante, un grupo de pacientes requiere dosis máximas inhaladas y alrededor de un 1% necesita de GC orales (asma corticodependiente). Una fracción pequeña va a tener resistencia a GC (refractarios o insensibles). Esta se define como ausencia de mejoría clínica o espirométrica después de tratamiento con altas dosis de GC (40 mg de prednisolona diaria por 15 días). La ausencia de respuesta espirométrica se define como la falta de mejoría de al menos un 15% en el FEV1 a los 15 días de tratamiento, ante la presencia de mejoría >15% con el uso de un broncodilatador de acción corta.14 Estos pacientes tienen una función pulmonar matutina más deteriorada, mayor grado de hiperreactividad bronquial e historia de asma familiar con mayor frecuencia que los asmáticos sensibles a GC. No tienen déficit de cortisol ni anormalidades en las hormonas sexuales y desarrollan los efectos secundarios de los GC, incluyendo el síndrome de Cushing.25
Alrededor de un 25% de los pacientes asmáticos fuman y tienen una respuesta reducida a los GC. El asma es más severa y hay una declinación más rápida de la función pulmonar en comparación con los asmáticos que no fuman.26 Los pacientes con EPOC también tienen poca respuesta a los GC. Este tratamiento no tiene efecto en la progresión de la enfermedad ni en la mortalidad; se ha reportado una potencial pero ligera reducción de las exacerbaciones,27 aunque recientemente este hallazgo ha sido cuestionado.28
En la EPOC, los GC no reducen células inflamatorias, citoquinas ni proteasas en el esputo, ni aún con GC orales. No solo no suprimen la inflamación neutrofílica de las vías aéreas, sino que inhiben su apoptosis.29,30 No obstante, alrededor de un 10% de los pacientes con EPOC sí responde a los GC inhalados. Estos pacientes poseen mayor número de eosinófilos en las vías aéreas, así como mayor reversibilidad con los broncodilatadores. Se ha planteado que en ellos es probable que exista, concomitantemente, asma.2
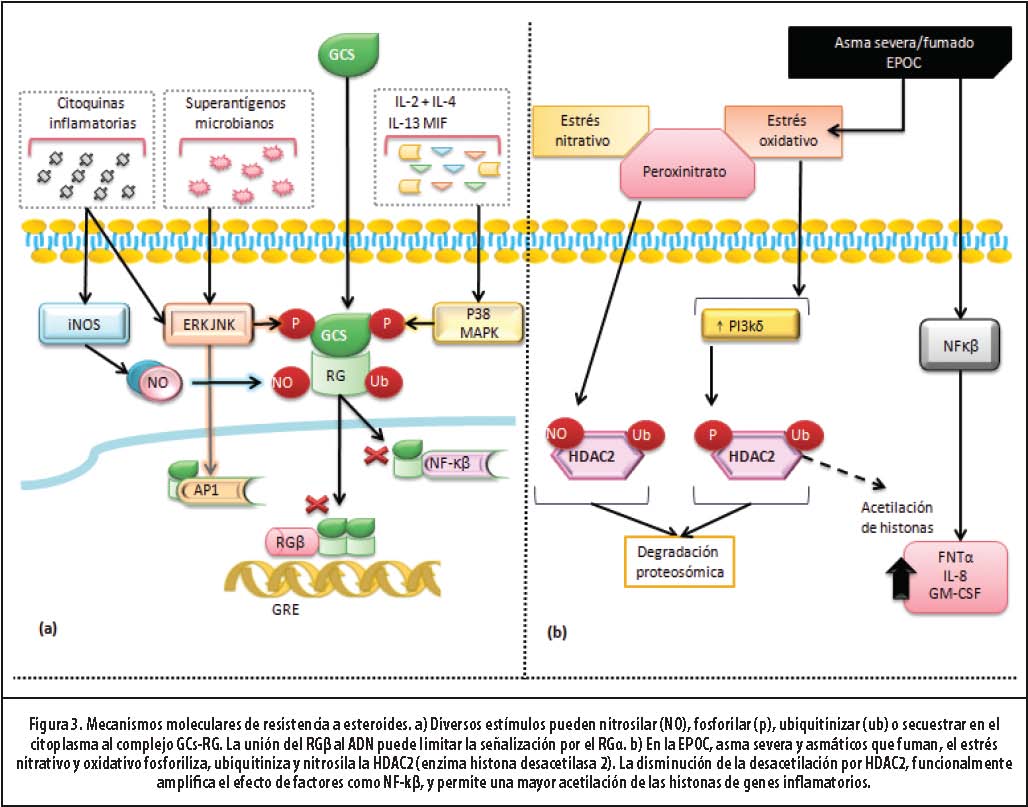
Mecanismos moleculares de resistencia
A continuación se detallan los mecanismos más conocidos de resistencia a GC. (Figura 3)
- Resistencia familiar a GC. El muy raro síndrome de resistencia familiar a los GC (solo una docena de casos han sido descritos) obedece a una mutación del RG. El resto de los pacientes con asma resistente a GC no tiene obvias anormalidades genéticas en la estructura del receptor.31
- Modificación del RG. Existe un subgrupo de pacientes resistentes que tiene aumentada expresión de IL-2, IL-4 e IL-13 en células T y monocitos. Estas activan la familia de las quinasas: p38 MAPK, JAK-3 (quinasa Janus-asociada-3) y JNK (quinasa c-jun N-terminal). Tales enzimas fosforilan los residuos de serina 226 o 211 del RG, y disminuyen su afinidad por los GC y la translocación nuclear.24,32,33,34 El MIF (factor inhibidor de la migración del macrófago) es una citoquina con potente acción antiglucocorticoide y su
actividad se ha encontrado aumentada en asma resistente a GC, lo que sugiere un rol potencial para terapias antiMIF.31
- Aumento de la expresión de RGβ. Se propone que este compite con la isoforma α por la unión al GRE o por la unión a moléculas coactivadoras. Dado que el RGβ no se une a los GC, no permite mediar su acción y se comporta como una forma dominante negativa. 13
- Aumento de la transcripción de factores proinflamatorios. La excesiva activación de AP-1 es otro mecanismo de resistencia. Esta proteína se une al RG y previene la interacción con el GRE y factores de transcripción. 35,36
![]() 5. Disminución de los linfocitos T reguladores. La IL-10 es una importante citoquina antinflamatoria e inmunorreguladora secretada por células T en respuesta a 36. Existen pacientes asmáticos GC resistentes que no secretan IL-10 en respuesta a la hormona y que al agregarles vitamina D3 (calcitriol), restauran la producción de la interleucina, lo que sugiere implicaciones terapéuticas. La vitamina D es un fuerte regulador del sistema inmune, particularmente en el control de células T regulatorias. Por lo tanto, un bajo ingreso en la dieta de la vitamina o poca exposición solar, podría contribuir a la resistencia a GC en enfermedades inflamatorias.37
5. Disminución de los linfocitos T reguladores. La IL-10 es una importante citoquina antinflamatoria e inmunorreguladora secretada por células T en respuesta a 36. Existen pacientes asmáticos GC resistentes que no secretan IL-10 en respuesta a la hormona y que al agregarles vitamina D3 (calcitriol), restauran la producción de la interleucina, lo que sugiere implicaciones terapéuticas. La vitamina D es un fuerte regulador del sistema inmune, particularmente en el control de células T regulatorias. Por lo tanto, un bajo ingreso en la dieta de la vitamina o poca exposición solar, podría contribuir a la resistencia a GC en enfermedades inflamatorias.37
Glucocorticoides en asma y EPOC/ Alvarado-González y Arce-Jiménez
6. Acetilación defectuosa de las histonas. Otro grupo de pacientes tiene acetilación defectuosa de la lisina 5 de la histona 4, y es resistente a dosis altas de GC inhalados, pero sufre los efectos secundarios de los GC.31
Implicaciones terapéuticas
La resistencia a los efectos antinflamatorios de los GC es una barrera mayor para el control efectivo de la enfermedad, en tanto implica gran morbilidad y altos costos de salud. Existen estrategias terapéuticas para tratar esta condición, y las más importantes se detallan de seguido.
- Esteroides disociados: los agonistas selectivos del RG (esteroides disociados) son más efectivos en la transrrepresión (efecto antinflamatorio obtenido al inactivar la acetilación de las histonas y estimular la desacetilación) que en la transactivación (media efectos secundarios, metabólicos y endocrinológicos).13 Varios GC disociados están en desarrollo y se investigan en la arena clínica, sin embargo, existe duda acerca de su eficacia.31 Si esto se lograra, se podrían obtener GC con amplio margen de seguridad, incluso para su uso oral, sin efectos adversos significativos. La reciente descripción de la estructura de cristal del RG podría ayudar al diseño de GC disociados.38
- Tratamientos antinflamatorios alternativos: no se ha encontrado que el tratamiento con inhibidores de calcineurina (p.ej., tacrolimus y ciclosporina A), efectivos en artritis reumatoide, sea eficaz en asma resistente.31
Drogas que estimulen la actividad de HDAC-2, como inhibidores de fosfodiesterasa 4 (p.ej., teofilina en dosis bajas), inhibidores de fosfoinositol-3-quinasa δ (PI3K-δ), antioxidantes e inhibidores de la óxido nítrico sintetasa inducible, pueden tener un papel promisorio en enfermedades resistentes a GC.39
Inhibidores de p38 MAPK están en desarrollo, particularmente en asma esteroide-resistente por hiperactividad de IL-2 e IL-4. No obstante, la toxicidad y sus efectos secundarios ( infección) han limitado su éxito.40 Lo mismo sucede con los inhibidores del NF-Kβ, por lo que solo están disponibles a nivel tópico.3
3. Reversión de la resistencia a esteroides: una opción atractiva para tratar la resistencia es revertir la causa. Por ejemplo, suspender el fumado en asmáticos que fuman.41 También se ha propuesto el uso de inhibidores de JNK, JAK-3 y de vitamina D3.42Inhibidores de MIF (moléculas pequeñas y anticuerpos monoclonales) están siendo explorados.33,43-46
La expresión y la actividad de la HDAC-2 se encuentran muy reducidas en macrófagos, vías aéreas y parénquima pulmonar de pacientes con EPOC, asmáticos fumadores y resistentes. El estrés nitrativo al formar peroxinitrito, nitrosila y los residuos de tirosina de la HDAC-2, conllevan a inactivación, ubiquitinación y degradación. El estrés oxidativo activa la PI3K-δ, la cual fosforila e inactiva la HDAC-231 y es común en muchas enfermedades inflamatorias severas. En EPOC, incluso cuando se haya suspendido el fumado, el estrés oxidativo persiste y, por lo tanto, la resistencia a los GC.13
Uno de los mecanismos de acción de teofilina es inhibir la PI3K-δ, que reduce el efecto del estrés oxidativo sobre HDAC-2 y permite aumentar la expresión y actividad de la enzima, lo que implica un efecto sinergístico antinflamatorio entre las dos drogas.
En conclusión, la resistencia a GC es importante en varias enfermedades inflamatorias comunes y complica su manejo clínico. Muchos mecanismos moleculares de este fenómeno han sido dilucidados, pero se requiere pruebas de laboratorio rápidas y de bajo costo para definir las vías involucradas. La investigación ha llevado a plantear nuevas estrategias terapéuticas, y en ese horizonte, tal vez a corto plazo se encuentre el tratamiento para estos pacientes.
Conflicto de intereses: ninguno de los autores tiene relación financiera con alguna entidad que tuviese interés en los resultados de este manuscrito.
Referencias
- Barnes P J, Adcock I M. How do corticosteroids work in asthma? Ann Intern Med 2003;139:359-70.
- Barnes P J. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol 2008;8:183-92.
- Barnes P J, Adcock I M. Glucocorticoid resistance in inflammatory diseases. Lancet 2009;373:1905-17.
- Barnes P J. Biochemical basis of asthma therapy. J Biol Chem 2011;286:32899-905.
- Barnes PJ, Adcock IM. Transcription factors and asthma. Eur Respir J 1998;12:221-34
- Busse WW, Lemanske RF, Jr. Asthma. N Engl J Med 2001;344:350-
- 62.
- Barnes P J, Chung K F, Page C. Inflammatory mediators of asthma: an update. Pharmacol Rev 1998;50:515-96.
- Adcock IM. Glucocorticoid-regulated transcription factors. Pulm Pharmacol Ther 2001;14:211-9.
- Hart L, Krishnan V, Adcock I M, Barnes P J, Chung K F. Activation and localization of transcription factor, nuclear factor-kappaB, in asthma. Am J Respir Crit Care Med 1998;158:1585-92.
- Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med 1997;336:1066-71.
- Adcock IM, Barnes PJ. Molecular mechanisms of corticosteroid resistance. Chest 2008;134:394-401.
- Bannister A, Schneider R, Kouzarides T. Histone methylation: dynamic or static? Cell 2002;109:801-6.
- Ogryzko V, Schiltz R, Russanova V, Howard B, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 1996;87:953-9.
- Ito K, Ito M, Elliott W M, Cosio B, Caramori G, Kon O M, et al. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N Engl J Med 2005;352:1967-76.
- Berger S. An embarrassment of niches: the many covalent modifications of histones in transcriptional regulation. Oncogene 2001;20:3007-13.
- Roth S, Denu J, Allis C. Histone acetyltransferases. Annu Rev Biochem 2001; 70:81-120.
- To M, Yamamura S, Akashi K, Charron C, Barnes P, Ito K. Defect of adaptation to hypoxia in patients with COPD due to reduction of histone deacetylase 7. Chest 2012;141:1233-42.
- Kagoshima M, Wilcke T, Ito K, Tsapruni L, Barnes P J, Punchard N, Adckock I M. Glucocorticoid-mediated transrepression is regulated by histone acetylation and DNA methylation. Eur J Pharmacol 2001;429:327-34.
- Jenuwein T. Translating the Histone Code. Science 2001;293:1074-80
- Ito K, Chung K F, Adcock I M. Update on glucocorticoid action and resistance. J Allergy Clin Immunol 2006;117:522-43.
- Anand R, Marmorstein R. Structure and mechanism of lysine-specific demethylase enzymes. J Biol Chem 2007;282:35425-9.
- ten Brinke A, Zwinderman A, Sterk P, Rabe K, Bel E. “Refractory” eosinophilic airway inflammation in severe asthma: effect of parenteral corticosteroids. Am J Respir Crit Care Med 2004;170:601-5.
- Barnes P J. Glucocorticosteroids: current and future directions. Br J Pharmacol 2011;163:29-43.
- Barnes P J. Mechanisms and resistance in glucocorticoid control of inflammation. J Steroid Biochem Mol Biol 2010;120:76-85.
- Thomson N, Chaudhuri R, Livingston E. Asthma and cigarette smoking. Eur Respir J 2004;24:822-33.
- Yang IA, Fong KM, Sim EH, Black PN, Lasserson TJ. Inhaled corticosteroids for stable chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2007:CD002991.
- Suissa S, Ernst P, Vandemheen K, Aaron S. Methodological issues in therapeutic trials of COPD. Eur Respir J 2008;31:927-33.
- Bourbeau J, Christodoulopoulos P, Maltais F, Yamauchi Y, Olivenstein R, Hamid Q. Effect of salmeterol/fluticasone propionate on airway inflammation in COPD: a randomised controlled trial. Thorax 2007;62:938-43.
- Culpitt S V, Rogers D F, Shah P, Matos D C, Russell E K R, E Louise, et al. Impaired inhibition by dexamethasone of cytokine release by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2003;167:24-31.
- Chikanza I, Kozaci D. Corticosteroid resistance in rheumatoid arthritis: molecular and cellular perspectives. Rheumatology 2004;43:1337-45.
- Marwick J, Adcock I M, Chung K F. Overcoming reduced glucocorticoid sensitivity in airway disease: molecular mechanisms and therapeutic approaches. Drugs 2010; 70:929-48.
- Yan Z-q, Hansson G. Innate immunity, macrophage activation, and atherosclerosis. Immunol Rev 2007;219:187-203.
- Irusen E, Matthews JG, Takahashi A, Barnes PJ, Chung KF, Adcock IM. p38 Mitogen-activated protein kinase-induced glucocorticoid
receptor phosphorylation reduces its activity: role in steroid-insensitive asthma. J Allergy Clin Immunol 2002;109:649-57.
- Miller A, Webb M, Copik A, Wang Y, Johnson B H, Kumar R, Thompson EB.p38 Mitogen-activated protein kinase (MAPK) is a key mediator in glucocorticoid-induced apoptosis of lymphoid cells: correlation between p38 MAPK activation and site-specific phosphorylation of the human glucocorticoid receptor at serine 211. Mol Endocrinol 2005;19:1569-83.
- Szatmáry Z, Garabedian M, Vilcek J. Inhibition of glucocorticoid receptor-mediated transcriptional activation by p38 mitogen-activated protein (MAP) kinase. J Biol Chem 2004;279:43708-
- 15.
- Bhavsar P, Hew M, Khorasani N, Torrego A, Barnes P J, Adcock I M, Chung K F. Relative corticosteroid insensitivity of alveolar macrophages in severe asthma compared with non-severe asthma. Thorax 2008;63:784-90.
- Lane S, Adcock I M, Richards D, Hawrylowicz C, Barnes P J, Lee T. Corticosteroid-resistant bronchial asthma is associated with increased c-fos expression in monocytes and T lymphocytes. J Clin Invest 1998;102:2156-64.
- Hawrylowicz C. Regulatory T cells and IL-10 in allergic inflammation. J Exp Med 2005;202:1459-63.
- Matthews J, Ito K, Barnes P J, Adcock I M. Defective glucocorticoid receptor nuclear translocation and altered histone acetylation patterns in glucocorticoid-resistant patients. J Allergy Clin Immunol 2004;113:1100-8.
- Bledsoe R K, Montana V G, Stanley T B, Delves C J, McKee D D, Consler T G, et al. Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell 2002;110:93-105.
- Dastidar SG, Rajagopal D, Ray A. Therapeutic benefit of PDE4 inhibitors in inflammatory diseases. Curr Opin Investig Drugs 2007;8:364-72.
- Medicherla S, Fitzgerald M F, Spicer D, Woodman P, Ma J Y, Kapoun A M, et al. p38alpha-selective mitogen-activated protein kinase inhibitor SD-282 reduces inflammation in a subchronic model of tobacco smoke-induced airway inflammation. J Pharmacol Exp Ther 2008;324:921-9.
- Chaudhuri R, Livingston E, McMahon A D, Lafferty J, Fraser I, Spears M, et al. Effects of smoking cessation on lung function and airway inflammation in smokers with asthma. Am J Respir Crit Care Med 2006;174:127-33.
- Xystrakis E, Kusumakar S, Boswell S, Peek E, Urry Z, Richard D F, et al. Reversing the defective induction of IL-10-secreting regulatory T cells in glucocorticoid-resistant asthma patients. J Clin Invest 2006;116:146-55.
- Hoi AY, Iskander MN, Morand EF. Macrophage migration inhibitory factor: a therapeutic target across inflammatory diseases. Inflamm Allergy Drug Targets 2007;6:183-90.
- Wada H, Kagoshima M, Ito K, Barnes PJ, Adcock IM. 5-Azacytidine suppresses RNA polymerase II recruitment to the SLPI gene. Biochem Biophys Res Commun 2005;331:93-9.