![]()
ISSN 0001-6012 eISSN 2215-5856
Acta Médica Costarricense 2025 / abril-junio; 67 (2): 1-10

Revisión narrativa sobre el síndrome de Takotsubo y la hemorragia subaracnoidea
(A Takotsubo syndrome and subarachnoid hemorrhage review)
Daniel Xu-Carranza1, Christian Ramírez Alfaro2, Carlos Alonso Lizano-Loría3
Afiliación Institucional:
1Caja Costarricense del Seguro Social. Hospital Monseñor Sanabria, Servicio de Medicina Interna, Puntarenas, Costa Rica. [email protected]
 0000-0003-0892-2037
0000-0003-0892-2037
2Caja Costarricense del Seguro Social. Hospital México, Unidad de Neurocríticos, servicio de Cuidado intensivo, San José, Costa Rica. [email protected]
0000-0001-7114-9396
3Caja Costarricense del Seguro Social. Hospital México, Unidad Coronaria, servicio de Cardiología, San José, Costa Rica. [email protected]
0009-0009-3827-9792
Abreviaturas:
ECG; electrocardiograma.
HSAa; hemorragia subaracnoidea aneurismática.
iECA; inhibidores de la enzima convertidora de angiotensina.
OTSVI; obstrucción del tracto de salida del VI.
STT; síndrome de takotsubo.
Fuentes de apoyo: Propia de los autores. No hubo participación económica por terceras partes.
Conflicto de intereses: No hay conflictos de interés por parte de los autores.
Contribución de los autores: Las contribuciones del autor primario se basaron en la planeación, recopilación de datos, análisis de la información y redacción. Las contribuciones del segundo autor fueron la planeación, recopilación de datos, revisión crítica y aprobación de la versión final.
Resumen
Los pacientes que presentan una hemorragia subaracnoidea aneurismática pueden llegar a desarrollar un síndrome de takotsubo como complicación cardiovascular concomitante. Esta complicación puede tener un comportamiento potencialmente letal o agregar una carga de morbilidad sustancial a la evolución del paciente. Por lo tanto, se considera un tema de relevancia y se realiza esta revisión con la intención de examinar los mecanismos fisiopatológicos, la presentación clínica, los estudios diagnósticos y el manejo de esta entidad.
Descriptores: cardiomiopatía de Takotsubo, síndrome del corazón roto, miocardiopatía de Tako-Tsubo, Obstrucción del Flujo de Salida Ventricular Izquierda, hemorragia subaracnoidea
Abstract
Patients with aneurysmal subarachnoid hemorrhage may develop takotsubo syndrome as a concomitant cardiovascular complication. This complication can be life-threatening or add a substantial burden of disease to the patient’s outcome. Therefore, this review is carried out with the intention of examining the pathophysiological mechanisms, clinical presentation, diagnostic studies and management of this entity.
Keywords: takotsubo syndrome, broken heart syndrome, stunned neurogenic myocardium, neurogenic stress cardiomyopathy, Takotsubo cardiomyopathy, Left Ventricular Outflow Obstruction, subarachnoid hemorrhage
Fecha de recibido: 30, setiembre, 2025 Fecha de aceptado: 02, diciembre, 2025
A lo largo de los años se ha documentado que distintos fenómenos patológicos a nivel encefálico pueden generar una serie de alteraciones cardiovasculares. Las primeras descripciones de estos fenómenos datan desde 1947, cuando se reportaron lesiones miocárdicas y eventos arrítmicos secundarios a enfermedades vasculares cerebrales. Desde entonces, esta interacción cerebro-corazón ha recibido múltiples nombres como: síndrome de Takotsubo (STT), síndrome del corazón roto, miocardio neurogénico aturdido, cardiomiopatía de estrés neurogénico.1,2 Independientemente de la entidad establecida, una vez instaurada la lesión cardiovascular, se dificulta el manejo de los pacientes neurocríticos y empobrece su pronóstico. En esta revisión nos centraremos en el STT como manifestación cardiaca secundaria a una hemorragia subaracnoidea aneurismática (HSAa).
El STT se describió formalmente en 1990 por Sato et al, es una cardiopatía aguda y transitoria, etiológicamente se puede dividir en dos grandes grupos: primaria y secundaria. Se entiende como primaria cuando ocurre en relación a un estrés psicológico o idiopático y secundaria cuando está en relación con un evento de estrés orgánico, como en asociación a la HSAa. El STT tiene tres características particulares: no tiene un defecto coronario culpable, es una cardiopatía reversible y las alteraciones morfológicas que ocurren en el miocardio no respetan la irrigación de las arterias coronarias.2,3
A pesar de tener más de 30 años desde su publicación sigue siendo una entidad poco reconocida y que una vez presentada complica el manejo de los pacientes con HSAa. Por lo tanto, se plantea la siguiente revisión bibliográfica con el objetivo de revisar y actualizar el manejo diagnóstico y terapéutico de esta patología cardiocerebral.
Metodología
Para la realización de esta revisión bibliográfica se realizó una búsqueda en las bases de datos de Medline, las bases de datos de la Universidad de Costa Rica por medio del SIBDI, la Biblioteca Nacional de Salud y Seguridad Social (BINASSS) y Google Scholar. Para la búsqueda se utilizaron las palabras claves: síndrome de takotsubo, hemorragia subaracnoidea aneurismática, aturdimiento miocárdico neurogénico y cardiomiopatía por estrés neurogénico. Se recopilaron artículos desde diciembre del 2006 hasta enero 2024 y primordialmente se incluyeron revisiones bibliográficas, revisiones sistemáticas, meta análisis y series de casos. Además, para la descripción clínica y diagnóstica, se revisaron estudios observacionales y revisiones sistemáticas, así como la revisión de guías clínicas para el análisis terapéutico.
Resultados
En el contexto epidemiológico, la exacta incidencia del síndrome de Takotsubo en HSAa es desconocida, esto se debe a que es una patología de reciente diagnóstico, con gran variabilidad de síntomas y presentación clínica. Distintos autores han reportado tasas de alrededor de 0.8-5%.4,5
Los factores de riesgo mayormente descritos para el desarrollo del STT posterior a una HSAa han sido: evento de alto grado (definida según las escalas de riesgo como Fisher, WFNS o Hunt-Hess) y sexo femenino.5 Con respecto a la edad, aunque generalmente se asocia a pacientes en edad post menopaúsica, se ha observado que los pacientes que desarrollan eventos neurológicos pueden presentar el STT más jóvenes.4
Fisiopatología
Actualmente no se han establecido los elementos fisiopatológicos exactos; no obstante, diversos mecanismos han sido propuestos y clásicamente se ha planteado el aumento excesivo de la actividad simpática a nivel cardiaco secundario a una ruptura aneurismática como mecanismo principal, la cual es capaz de generar una liberación masiva de catecolaminas responsables de inducir injuria miocárdica.1, 6 Sin embargo, al día de hoy se han descrito otras vías fisiopatológicas plausibles (más allá que el efecto singular de las catecolaminas sobre el miocardio). Se han considerado mecanismos microvasculares coronarios, hormonales e inflamatorios; la interacción cerebro – corazón ha demostrado ser más compleja que la simple liberación de epinefrina y noreprinefrina.7
Como muestra la figura 1, la ruptura aneurismática produce una injuria directa, a nivel encefálico, secundaria a procesos de isquemia, hemorragia, efecto de masa por el hematoma e inflamación local. Estas lesiones, a su vez, producen cambios homeostáticos y vasculares regionales, como: alteración del flujo sanguíneo cerebral, lesión de la barrera hematoencefálica, cambios en la presión de oxígeno, dióxido de carbono, liberación de neurotransmisores (p. ej. Glutamato, adenosín trifosfato ATP) y moléculas inflamatorias.8,9 Además, las lesiones aneurismáticas suelen localizarse en proximidad con el hipotálamo, tallo e ínsula; regiones que se encuentran relacionadas con el control autonómico. Existen dos centros destacables en el control del tono simpático a nivel de SNC: el núcleo paraventricular (NPV) en el hipotálamo y la médula rostral ventrolateral (MRVL) en el bulbo raquídeo. Estos núcleos envían sus eferencias al núcleo intermedio lateral (NIL), ubicado en la médula espinal, de donde parte la primera neurona responsable de la conducción eferente simpática, su axón viaja a los ganglios para y prevertebrales y de ahí al resto de sistemas y órganos.8,9 Por medio de las lesiones descritas, la ruptura de un aneurisma en contigüidad con estos territorios puede desencadenar en una respuesta catecolaminérgica con extensión sistémica al estimular el eje del NPV-MRVL-NIL. Al mismo tiempo, esta respuesta autonómica estimula las glándulas suprarrenales que inducen la liberación de cortisol y catecolaminas. Esto amplifica y perpetúa las lesiones sistémicas y cerebrales, creando así una retroalimentación positiva entre el SNC y los órganos periféricos mediante una comunicación neuro-endocrina del tallo cerebral con la glándula suprarrenal (encargada de la regulación simpática por medio de la liberación de norepinefrina y epinefrina).9,10 Justamente, se ha demostrado que los pacientes con HSAa presentan niveles séricos elevados de catecolaminas.1,10
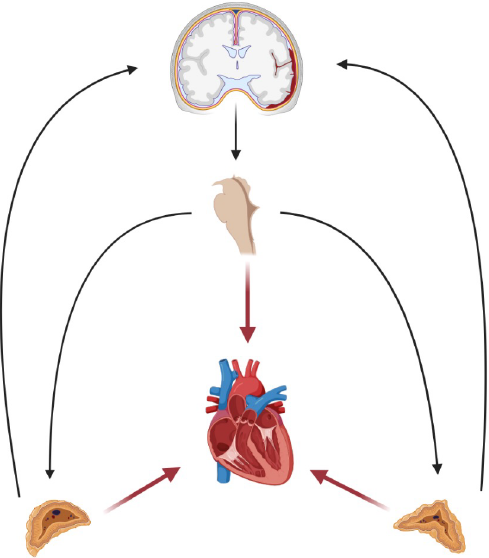
Figura 1. Con la ruptura aneurismática se da la injuria encefálica y estimulación de centros simpáticos a nivel del SNC y periférico (glándula suprarrenal). Las flechas negras representan el eje neuro-endocrino y su retroalimentación positiva. Mientras que las flechas rojas representan la descarga adrenérgica central y periférica sobre el corazón.
La razón por la cual la epinefrina y la norepinefrina producen disfunción cardiaca tiene que ver con el tipo de receptor que activan y la distribución de los mismos. Tanto la norepinefrina como la epinefrina actúan sobre el miocardio gracias a la interacción que tienen con los receptores β. En el miocardio podemos encontrar receptores β1 y β2, los cuales tienen una distribución específica a lo largo del músculo cardiaco y una interacción particular con cada catecolamina. La epinefrina actúa a través de receptor β2, a niveles fisiológicos tendrá un efecto inotrópico positivo por su interacción con la proteína Gs, mientras que a nivel suprafisiológico tendrá un efecto inotrópico negativo debido al cambio molecular en la proteína G, lo cual conlleva a acoplarse a una Gi. Estos receptores se encuentran preferencialmente ubicados en el ápice. Lo contrario sucede con la NE que estimula los receptores β1 que se encuentran en mayor cantidad a nivel del miocardio basal y cuya estimulación conduce a un efecto inotrópico positivo.
El cambio en la proteína G acoplada a los receptores adrenérgicos beta es un fenómeno específico de la epinefrina. Esta distribución también explica la morfología clásica (>80% de los casos) del STT caracterizada por un abalonamiento apical (figura 2). En los casos de las morfologías atípicas, se ha planteado que la activación de los receptores β2 produce que estos se incorporen a nivel intracelular (captados por vesículas lipídicas) lo cual genera menor disponibilidad de los mismos y un cambio en la distribución usual de receptores β1 y β2. 2,3,10
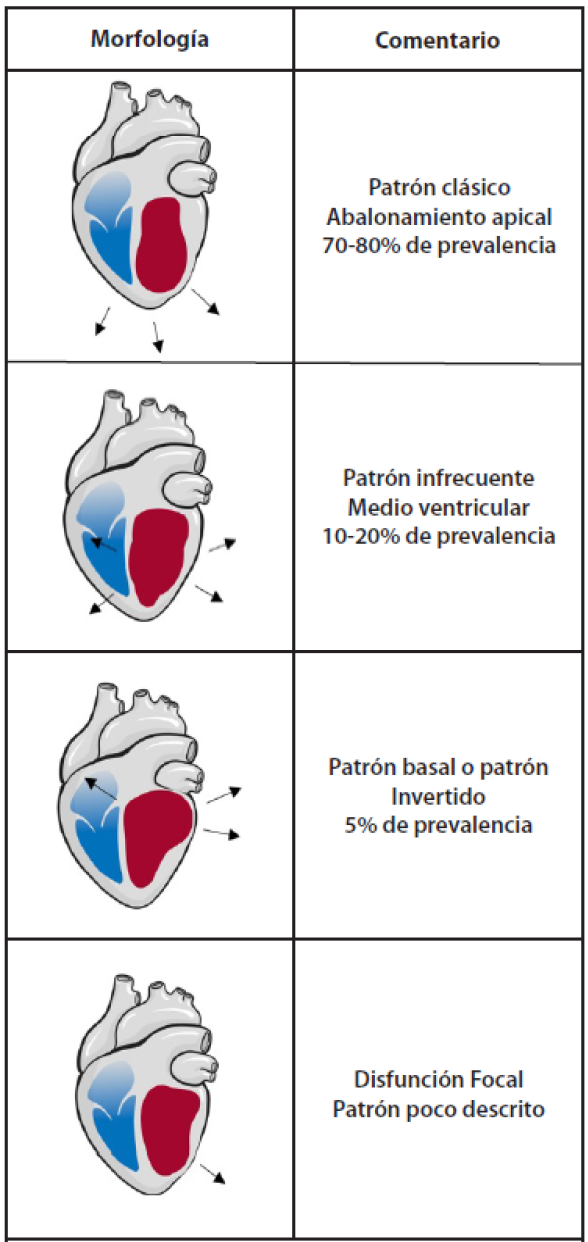
Figura 2. Patrones ecocardiográficos en relación con el Síndrome de Takotsubo.11,22
La evidencia actual respalda el rol de las catecolaminas en el desarrollo fisiopatológico del STT. Esto se ha demostrado a partir de estudios patológicos donde se ha documentado evidencia de bandas de necrosis miocárdicas con estados catecolaminérgicos elevados; presencia de alto niveles séricos de norepinefrina de forma sistémica y regional cardiaca. Además, de la reproducibilidad clínica del STT cuando hay estimulación de receptores beta de forma experimental y estudios nucleares que documentan una menor recaptura postsináptica simpática y por lo tanto, mayor exposición a catecolaminas.10
Por otro lado, se han propuesto otros mecanismos además de la acción directa de las catecolaminas, como por ejemplo: la presencia de alteraciones vasculares, metabólicas, inflamatorias y genéticas.Además, se han descrito vías fisiopatológicas asociadas al STT. Estudios iniciales habían planteado la posibilidad de cambios coronarios como responsables del STT.10 Actualmente, se ha descartado la hipótesis de un proceso obstructivo epicárdico en el desarrollo fisiopatológico del STT, esto gracias a estudios angiográficos con uso de ultrasonido intravascular (IVUS) o tomografía por coherencia óptica (OCT).10,11 No obstante, con respecto a la microcirculación coronaria, la evidencia es más controversial, sin lograr determinar el rol exacto en la enfermedad. Se ha documentado en estudios de provocación farmacológica que la presencia de disfunción microvascular y su recuperación puede cambiar el pronóstico del STT. Por lo tanto, se ha propuesto que este puede ser un mecanismo coadyuvante en ciertos pacientes, aunque probablemente no con un papel central.1 Además, el comportamiento de la troponina y los territorios comprometidos (que no suelen corresponder a una región coronaria específica) tampoco sugieren una causa isquémica como mecanismo principal.
Así mismo, estudios con emisión de positrones con fluorodesoxiglucosa y tomografía computarizada por emisión de fotón único sugieren anomalías en el metabolismo mitocondrial, esto debido a la reducción en el consumo de glucosa, reducción en el metabolismo de ácido grasos y la acumulación de lípidos intracelulares. Se estima que estas alteraciones son secundarias al efecto indirecto de las catecolaminas, a la disfunción microcirculatoria y a un componente inflamatoria local.10 Con respecto a este último, estudios patológicos han documentado infiltraciones celulares por polimorfonucleares y macrófagos a nivel miocárdico, asociado además a la presencia de edema apical en imágenes de resonancia magnética.10 Por último, biopsias adquiridas en la fase aguda del STT han reportado alteraciones genéticas en los genes reguladores del manejo de calcio intracelular, específicamente una regulación a la baja del gen SERCA2 y una regulación al alta de la sarcolipina. Mecánicamente esto se refleja con una disfunción tanto en la contracción como en la relajación del músculo cardiaco.10
Finalmente, a pesar de que se han expuesto las diversas vías de forma separada, se debe entender el proceso fisiopatológico como un evento integral en el cual, posterior a un evento primario como una HSAa, los diversos estímulos adrenérgicos, inflamatorios, metabólicos y genéticos trabajan de forma conjunta en una misma vía patológica para acabar en un cuadro clínico congruente con STT.
Presentación clínica y evaluación
Durante una HSAa la injuria cardiaca puede ser evidente o desarrollarse horas después de la ruptura aneurismática. Clásicamente los pacientes se aquejan de dolor torácico agudo, disnea o síncope. Las complicaciones se expresarán en un espectro de severidad clínica, algunos pacientes cursarán asintomáticos, mientras que otros desarrollarán insuficiencia cardiaca y edema pulmonar.12, 13 En su espectro más severo algunos podrán presentar choque cardiogénico, eventos arrítmicos o arresto cardiaco. La incidencia que se ha descrito de los principales síntomas en pacientes con injurias cerebrales son: dolor torácico (63-90%), disnea (18-32%), náusea y vómitos (16%), síncope (3-4%), edema pulmonar (40%) y arresto cardiaco (1-2%).5, 13
Se debe tomar en cuenta que los pacientes con HSAa no siempre serán capaces de reportar síntomas dado la alteración en el estado de consciencia. Por lo tanto, la sospecha diagnóstica surgirá de la tendencia clínica y hemodinámica del paciente, así como de los cambios electrocardiográficos y de los biomarcadores. 12, 14
La instauración de la insuficiencia cardiaca o choque cardiogénico en la HSAa es secundaria a dos fenómenos: disfunción sistólica y diastólica del ventrículo izquierdo (VI) y/u obstrucción dinámica del tracto de salida del VI (OTSVI).15 Independientemente de cuál sea el mecanismo, la hipotensión y la disminución de la oxigenación puede ser particularmente devastadora durante el periodo pico de vasoespasmo cerebral. Tanto el índice cardiaco deprimido como la presencia de complicaciones pulmonares en HSA son predictores de mayor mortalidad y peores desenlaces neurológicos.16-18
Además de las complicaciones hemodinámicas en relación a la función sistólica y diastólica del VI, existen otras dos complicaciones clínicas relevantes en los pacientes con STT que son la presencia de arritmias y la formación de trombos intracavitarios.9,11 Las arritmias más frecuentemente encontradas serán la taquicardia sinusal y la fibrilación auricular (FA).13 No obstante, también se puede presentar taquicardia o fibrilación ventricular o así como bloqueos atrio-ventriculares (BAV).
En el contexto hospitalario, se dispone de varios recursos complementarios para la evaluación clínica del paciente, como se describe a continuación:
1. Electrocardiograma (ECG): las alteraciones en el ECG ocurren en un 60-90% de los pacientes con ictus.12 Estas anormalidades se pueden organizar como cambios en la morfología o en el ritmo del ECG. Estos cambios suelen ser transitorios, inespecíficos y tienden a variar en el tiempo. Las irregularidades morfológicas más reportadas son las desviaciones del segmento ST, inversiones de la onda T y prolongación del intervalo Qt corregido.19,20 Se han descrito tres estadios distintos en cuanto a estos cambios electrocardiográficos: 21 Estadio 1: desviaciones en el ST en las primeras horas del inicio de los síntomas. Estadio 2: inversión de la onda T y prolongación del QTc de varios días de duración. Estadio 3: resolución de los cambios electrocardiográficos que puede tomar varios meses. Así mismo, puede haber trastornos del ritmo como bradicardia y taquicardia sinusal, FA, BAV y taquiarritmias ventriculares o supraventriculares.17 Todos estos cambios se suelen ver predominantemente en las primeras horas la HSAa.
2. Biomarcadores: Por una parte, la troponina se suele elevar en la mayoría de los pacientes con HSA.19 Sin embargo, no suele llegar a niveles altos y es más frecuente verla elevada en HSA severas (según la escala de Hunt y Hess), con un pico mayor en el día del ictus.20
Por otra parte, el péptido natriurérico tipo B (BNP) se suele elevar más frecuentemente y en mayor cantidad que la troponina. Esta se ha asociado a anormalidades contráctiles, fracción de eyección (FE) reducida, disfunción diastólica, edema pulmonar, elevación de troponina, así como aumento de mortalidad.6,16,17
La elevación de biomarcadores cardiacos tiene una correlación pronóstica y se suele asociar con peores desenlaces clínicos (lesión neurológica más severa y mortalidad), así como mayor disfunción ventricular, edema de pulmón e inestabilidad hemodinámica.16
3. Ecocardiografía (ECO): la ecocardiografía usualmente será el primer método de imagen que utilizaremos en la evaluación de la morfología funcional cardiaca de los pacientes con sospecha de STT (figura 2). Este, por su modalidad no invasiva puede llevarse a cabo al pie de la cama del paciente ya que es reproducible. Cuando se realiza un ECO en un paciente con sospecha de STT, se procura la evaluación de a) la morfología al permitir la identificación de la variante del STT, especialmente el balonamiento apical y medio ventricular, ambas con un patrón circunferencial (figura 3), pues las otras formas de STT son más difíciles de documentar en la ecocardiografía; b) la función sistólica y diastólica pues de forma tradicional se puede utilizar la fracción de eyección (FE) y la E/e’ respectivamente;22 aunque más actualizada, para facilitar esta evaluación, se está recurriendo al uso del “speckle tracking”, herramienta ecocardiográfica que permite valorar la deformación longitudinal, circunferencial y radial del miocardio;22 y c) las complicaciones mecánicas, siendo la más común la OTSVI, que puede acompañarse de un movimiento septal anterior (SAM) de la válvula mitral asociado a regurgitación mitral (RM). Entre otras complicaciones están: ruptura miocárdica, RM no asociada a la OTSVI y la formación de trombos intracavitarios.22
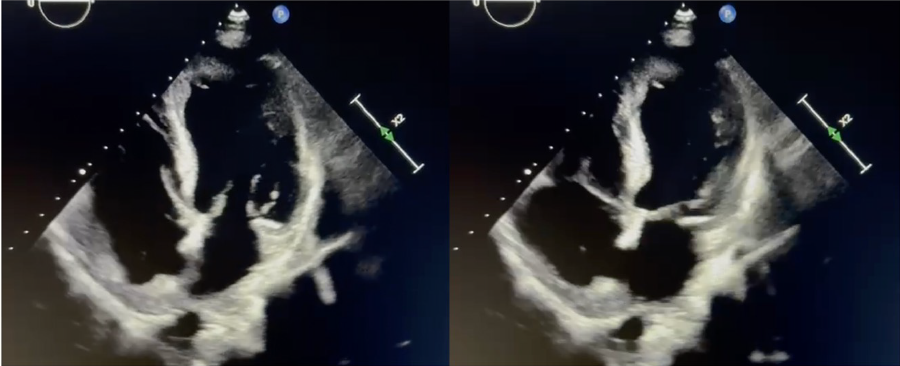
Figura 3. Evaluación ecocardiográfica del Síndrome de Takotsubo. Imagen izquierda en diástole e imagen izquierda en sístole con la morfología típica del abalonamiento apical, esto es observado en más del 80% de los casos.22
4. Coronariografía: es una prueba indispensable para el diagnóstico y pronóstico en la fase aguda ya que el principal diagnóstico diferencial del STT es el SCA (síndrome coronario agudo). Con la coronariografía se puede descartar lesiones coronarias significativas que podrían justificar el cuadro clínico. Es importante señalar que las lesiones coronarias obstructivas pueden coexistir con el STT. No se trata de documentar ausencia de lesiones angiográficas, sino de la congruencia que tengan estas lesiones con las anormalidades en la morfología cardiaca, ya que en el STT la lesión miocárdica se extiende más allá de la irrigación coronaria.22 Además, se pueden usar otras técnicas como el IVUS o el OCT para valorar la presencia de infarto miocárdico no obstructivo (MINOCA).23 Por último, la ventriculografía puede identificar alteraciones en la pared miocárdica sugestivas de STT y otras complicaciones como la regurgitación mitral, trombos intracavitarios y la medición de presiones ante la sospecha de OTSVI.22
5. Angiotomografía: se ha propuesto el uso de la misma para evaluar lesiones coronarias en STT. Tiene la ventaja que no es invasiva, por lo que en poblaciones seleccionadas (estables, sin elevación del segmento ST o dolor torácico persistente) podría ser una alternativa a la angiografía.23 Aunque la ausencia de lesiones angiográficas favorece el diagnóstico de STT se debe recordar que la presencia de enfermedad coronaria no descarta el STT. Siempre se debe correlacionar las lesiones coronarias con la extensión de la disfunción miocárdica.
6. Cardiorresonancia magnética (RMN): es considerada la prueba ideal para el STT y para descartar otras sospechas diagnósticas como el IAM, al demostrar la ausencia de necrosis miocárdica en las imágenes a través de ausencia de realce tardío de gadolinio.22 Por otra parte, es específico para el STT el edema miocárdico en los segmentos afectados del VI en la secuencia T2.23 El edema se resuelve en el seguimiento, con normalización de la función sistólica ventricular izquierda y de los cambios electrocardiográficos.
Como un insumo previo para el diagnóstico, la información aportada mediante la evaluación permite hacer una clasificación del STT según los cambios morfológicos del miocardio vistos por ECO (figura 2); se describen principalmente cuatro patrones11 , aunque la forma típica (apical ballooning) es la más frecuente (figura 3), afecta al ápex y a los segmentos medioventriculares, estos se encuentran hipocinéticos o discinéticos y con hipercontractilidad compensatoria de los segmentos basales (81,7% de los pacientes). Le sigue la forma medioventricular que afecta los segmentos medios del VI (14,6%);24 la forma basal que afecta los segmentos basales del VI con hipercontractilidad del ápex (2,2%);25 y la forma focal, con compromiso principal de un solo segmento del VI, como la cara lateral o la inferior, o del ventrículo derecho (VD) (1,5%).11
Diagnóstico
El diagnóstico del STT durante la presentación inicial es un reto y muchas veces se necesitará de estudios invasivos para la confirmación del mismo. Esto se debe a que sus manifestaciones clínicas se asemejan en gran medida al síndrome coronario agudo o la miocarditis. El diagnóstico del STT se obtendrá confirmando que el deterioro en la función cardiaca se extiende más allá de la perfusión coronaria por un vaso en particular, así como la reversibilidad del mismo.20, 26 Para demostrar dichos requisitos se hará uso de las herramientas clínicas ya descritas (historia clínica, ECG, biomarcadores y estudios de imágenes).
Adicionalmente, se han propuesto diferentes criterios clínicos con la intención de determinar el riesgo pretest para el diagnóstico del STT.20 A lo largo del tiempo se han utilizado los criterios de Gothenberg, los criterios italianos, los de la Clínica Mayo, los basados en resonancia magnética (RM) y, por último, los criterios InterTak definidos en el 2018. Estos últimos han sido recomendados tanto por entidades norteamericanas como europeas para el abordaje diagnóstico de pacientes con sospecha de STT.21,2. Los criterios de InterTak se muestran en el cuadro 1, la sumatoria de los puntajes tiene como meta alcanzar más de 70 puntos para poder atribuir el cuadro clínico al síndrome de Takotsubo.20, 27
La sospecha inicial del STT dependerá del reporte de síntomas junto con la monitorización del paciente. Una vez establecida la duda diagnóstica se recomienda hacer un “triage” según el ECG.28 En ausencia de elevación del punto J o segmento ST, el estado clínico del paciente y su perfil hemodinámico dictarán las medidas a seguir. En caso de pacientes estables la evaluación se podrá concretar con el uso de una escala de riesgo (interTAK) (cuadro 1) y el ECO. Si estas son congruentes con un STT, se podrá corroborar el diagnóstico con un angiografía coronaria o control ecocardiográfico según la clínica del paciente. De lo contrario, se deberá indagar en los posibles diagnósticos diferenciales. Así mismo, pacientes con elevación del punto J o segmento ST y con choque cardiogénico debería de evaluarse angiográficamente con prioridad. Si bien es cierto se puede hacer uso de la RMN o la tomografía de coronarias, estos son estudios no tan accesibles en la mayoría de los centros hospitalarios. En un futuro probablemente se podrán incorporar estas herramientas al abordaje diagnóstico.
|
Cuadro 1. Criterios de riesgo según la escala InterTak. La sumatoria del puntuaje alcanza 70 o más puntos para atribuir el cuadro clínico al Síndrome de Takotsubo.20, 27 |
|
|
Criterios |
Puntuaje |
|
Mujer |
25 |
|
Evento emocional |
24 |
|
Injuria orgánica |
13 |
|
Ausencia de depresión de ST |
12 |
|
Desorden psiquiátrico |
11 |
|
Desorden Neurológico |
09 |
|
Prolongación del QT |
06 |
|
Corte Diagnóstico: |
|
|
>70 puntos: Síndrome de Takotsubo (95% de especificidad) |
|
Manejo del Síndrome de Takotsubo
Los pacientes que presentan una HSAa y que desarrollan un STT pueden plantear un dilema terapéutico, por un lado, tenemos el compromiso neurológico y, por otro lado, un paciente con disfunción cardiaca. No existen lineamientos oficiales sobre el manejo médico de estos pacientes, dada la ausencia de ensayos clínicos.28 Por lo tanto, las indicaciones terapéuticas deben ir dirigidas a la individualidad fisiopatológica de las anomalías cardiacas y cerebrales. A continuación, se revisará el manejo propuesto hasta la fecha para esta entidad.
Como parte del manejo, una vez identificado el STT los pacientes deberán someterse a un periodo de monitorización continua del trazo electrocardiográfico, valoración del QTc y constantes vitales. Esto ante el riesgo de arritmias y el deterioro hemodinámico y/o respiratorio.
A continuación, debemos determinar (concomitantemente aseguradas las medidas de neuroprotección) el grado de severidad cardiogénica. El STT por su naturaleza evolucionará en la mayoría de los casos hacia la recuperación, este desenlace puede tardar unas cuantas semanas. Al tener en cuenta sus posibles espectros de complejidad, nos apoyaremos en el estado hemodinámico, el compromiso respiratorio y la evaluación ecocardiográfica para definir la gravedad y clasificaremos al paciente en tres grupos clínicos: i. Pacientes con inestabilidad hemodinámica, es decir, choque cardiogénico. ii. Pacientes con disfunción ventricular y clínica de insuficiencia cardíaca. Iii Pacientes con datos de injuria miocárdica asintomáticos.
En este punto, se debe individualizar a los pacientes que se beneficiarán de un monitoreo hemodinámico semiinvasivo o invasivo. El seguimiento y la vigilancia es la norma en aquellos pacientes con STT sin insuficiencia cardiaca, aunque se podría considerar el uso de un inhibidor de la enzima convertidora de angiotensina (iECA), un (antagonista del receptor de angiotensina II) ARA II o un β-bloqueador.2 Sin embargo, en pacientes que desarrollan insuficiencia cardiaca o estado de choque el manejo probablemente requerirá de mayores intervenciones y monitoreo. Para definir las mismas es importante identificar si hay presencia de OTSVI, esta se puede presentar hasta en 20% de los pacientes con STT.28, 29 Aunque la clínica define la estrategia de manejo, reconocer la presencia de la OTSVI es fundamental para la toma de decisiones terapéuticas.29 Esto lo podemos realizar a partir del uso de la ECO transtorácica o los datos de la ventriculografía.20
En relación con la OTSVI, definida como la presencia de un gradiente intraventricular > 30mmHg (hemodinamicamente significativo cuando es mayor de 50mmHg).30 Este componente obstructivo es causado por el estado hipercinético de los segmentos basales del miocardio que pueden generar un desplazamiento hacia el interior de la válvula mitral conocido como movimiento sistólico anterior (SAM). Este se puede generar por dos mecanismos: un aumento en la velocidad del flujo sanguíneo a través del tracto de salida del VI que genera un fenómeno de Venturi y que desplaza hacia anterior la válvula mitral; o bien, la válvula se puede desplazar por el cambio de la dirección del flujo sanguíneo intracavitario dado el aumento de la resistencia al flujo de salida.30 Este fenómeno se da predominantemente en los pacientes con morfología típica, más del 80% de los pacientes con STT y por tanto, es esencial recordar que las medidas terapéuticas clásicas usadas en pacientes con insuficiencia cardiaca o estado de choque (diuréticos, vasopresores, inotrópicos, vasodilatadores) pueden deteriorar el estado clínico de aquellos pacientes con obstrucción.2
Como entre un 12 y 45% de los pacientes que presentan STT pueden llegar a desarrollar insuficiencia cardiaca aguda y estado de choque,20 se describen a continuación las medidas recomendadas para estas complicaciones. Primero, en caso de pacientes con choque cardiogénico, los esfuerzos terapéuticos se centrarán en optimizar la función contráctil y el gasto cardiaco, para ello se debe determinar si hay OTSVI; en cuya presencia la meta en estos pacientes es disminuir el estado hipercinético de los segmentos basales y evitar los gradientes abruptos de presión entre el ventrículo y la aorta. Esto último se obtiene optimizando las presiones de llenado y aumentando la poscarga. Se debe evitar el uso de fármacos inotrópicos, así como de vasodilatadores y diuréticos, ya que disminuyen la precarga y la poscarga, elevando así el gradiente de presiones intracavitarias y amplificando el efecto hipercinético basal.30 Las medidas médicas a priorizar serían optimizar la precarga con adecuada resucitación, disminuir la dosis de inotrópicos o considerar su suspensión, valorar el uso de β-bloqueadores de corta acción en ausencia de bradicardia, FE reducida y QTc > 500ms.28 Además, en caso de persistir con hipotensión a pesar de estas medidas se puede probar el uso de presores sin efecto inotrópico como la vasopresina o la fenilefrina.
En ausencia de OTSVI, será un choque cardiogénico sin obstrucción del tracto de salida, la meta será recuperar la función contráctil. Para ello se prefiere el uso de inodilatadores no catecolaminérgicos, en respuesta a la fisiopatología adrenérgica del STT. Las medidas farmacológicas más recomendades son levosimendan, inotrópico favorito debido a que produce un menor consumo miocárdico de oxígeno, para aquellos pacientes con disfunción sistólica y sin OTSVI, en infusión continua por 24 horas a una dosis de 0.1 ug/kg/min.7,11 En caso de persistir el deterioro se podría valorar agregar vasopresores (norepinefrina o fenilefrina) a las dosis más bajas posibles o incluso utilizar otros inotrópicos como dobutamina.11
Si a pesar de todas estas medidas el paciente continúa con deterioro hemodinámico, se deberá valorar la colocación de soporte circulatorio mecánico.30,34 Actualmente se cuenta a nivel nacional con balón de contrapulsación aórtico (BCPA), dispositivos de asistencia ventricular y terapia de oxigenación por membrana extracorpórea (ECMO). Estas terapias se suelen considerar como un puente hacia la recuperación en vista de la naturaleza reversible y transitoria del STT.7 No obstante, se debe tener en cuenta que la presencia de OTSVI contraindica la colocación del BCPA, esto debido a que la disminución de la poscarga puede empeorar el gradiente ventrículo/arterial del VI y amplificar el movimiento dinámico obstructivo del miocardio basal.30
Por otra parte, cuando los pacientes que desarrollan insuficiencia cardiaca pero se mantiene hemodinámicamente estables, la terapia se dirigirá a reducir las presiones del llenado del VI y así aliviar la sobrecarga retrógrada de presiones, permitir una adecuada disminución del consumo miocárdico de oxígeno, mejorar la perfusión coronaria y optimizar la contractibilidad miocárdica al desplazar la curva de Frank Starling.20 Para ello se puede hacer uso de la combinación de iECAS, ARA II, diuréticos, vasodilatadores venosos o arteriales (p. ej nitroglicerina, hidralazina/isosorbide) y β-bloqueadores.
Siempre se debe mantener un adecuado monitoreo y vigilancia de variables hemodinámicas, en caso de sospechar de una posible OTSVI se debe considerar el uso betabloqueadores y evitar las reducciones en la poscarga con la intención de mejorar los gradientes de presión intraventriculares. En estos casos se recomienda mantener una adecuada hidratación y el uso de beta-bloqueadores con vida media corta en infusión continua como el esmolol.30, 32 No obstante, en nuestro medio no contamos con dicha opción farmacológica, por lo que otros autores han utilizado propanolol IV a una dosis de 0.1 mg/kg/día sin sobrepasar los 4 mg máximos.2 El argumento a favor del uso de los β-bloqueadores se basa en sus efectos farmacológicos, ya que ofrecen una contraposición a la exposición masiva de catecolaminas, optimizan el requerimiento energético del miocardio, puede disminuir el riesgo de arritmias y la obstrucción del tracto de salida.31, 32 Es importante recalcar que ante el uso de infusiones de β-bloqueadores se debe dar seguimiento cuidadoso a estos pacientes, y con aquellos que no mejoren y se deterioren clínicamente se deberá suspender la infusión.
Recientemente se ha propuesto el uso de insulina asociado con los β-bloqueadores. El fundamento fisiopatológico de esta combinación se basa en los efectos sinérgicos de la insulina con los beta-bloqueadores de corta acción, al destacar el aumento de la contractibilidad por parte de la insulina y su oposición a los efectos de las catecolaminas sobre el miocardio (alcanzados a altas dosis de insulina).31 No obstante aún faltan mayores estudios clínicos y evidencia robusta que respalden dicha intervención.
Por otra parte, la formación de trombos apicales y el riesgo de embolizaciones subsiguientes se ha planteado como una posible complicación del STT. Suele aparecer en 1-9% de los pacientes que desarrollan STT.2,31 Sin embargo, no se recomienda la anticoagulación de forma profiláctica; aunque se puede considerar en aquellos pacientes con grandes áreas aquinéticas o con una FE < 30%.28 En caso de documentarse la formación de un trombo apical, se ha sugerido la anticoagulación por al menos 3 meses o hasta que se recupere la función contráctil.20
Las arritmias pueden ocurrir en un 25% de los pacientes, entre ellas la más común es la fibrilación atrial y ocurre hasta 9% de los pacientes durante los primeros días de instaurado el STT.20 De forma concomitante, estos pacientes pueden presentar prolongación del intervalo QT, este hallazgo es un factor de riesgo para el desarrollo de trastornos del ritmo.20 Entonces, se ha recomendado el uso de beta-bloqueadores como posible protección contra la aparición de arritmias. Sin embargo, se debe evitar en pacientes con QT prolongado, bradicardia o BAV.2,31 También se debe optimizar el control hidroelectrolítico y asegurar los adecuados niveles de potasio y magnesio. Una vez establecida la disritmia se deberá continuar el manejo recomendado por las guías clínicas, ya sea farmacológico, eléctrico o con el uso de dispositivos transitorios o permanentes (p. ej. marcapasos).
En cuanto al manejo a largo plazo, se ha recomendado el uso de iECAS y ARA II ya que estos tienen un beneficio discreto en cuanto a mortalidad en el primer año postevento, por lo tanto, actualmente se recomienda mantenerlos crónicamente.31 Contrariamente, los β-bloqueadores no demostraron este impacto, aunque se ha sugerido su continuación crónica debido a sus mecanismos plausibles sobre la modulación simpática para disminuir el riesgo de recurrencia.11
La aspirina y las estatinas deberían de indicarse en aquellos pacientes que se beneficien de prevención secundaria, su rol de forma crónica no ha sido recomendado y no ha demostrado impacto en recurrencia o eventos cardiovasculares. No obstante, durante la fase aguda algunos estudios demostraron menor incidencia de eventos cardiovasculares con el uso de terapia antiplaquetaria, por lo tanto, algunos autores han abogado por el uso de aspirina en periodos cortos.7, 32
Finalmente, el pronóstico de los pacientes con STT se ha ido modificando con la recolección de la información. Inicialmente se asociaba al STT con un pronóstico favorable en vista de su cualidad de “reversibilidad”; sin embargo, se ha documentado que la mortalidad intrahospitalaria del STT alcanza 4-6%.20 Además, a largo plazo se ha notado un aumento en la morbimortalidad de los pacientes, sea morbilidad no cardiogénica como eventos cerebrovasculares y mayor predisposición a malignidad. Así mismo, se ha observado persistencia de afectación cardiaca, como edema y reservas enérgicas disminuidas, aun cuando se haya recuperado la fracción de eyección.10, 33
En consecuencia, de forma general es aconsejable un manejo interdisciplinario de los pacientes que desarrollan complicaciones cardiovasculares, acompañado por por especialistas en cardiología, neurocirugía y cuidado crítico; 34 que facilite una intervención expedita del aneurisma intracraneal, teniendo siempre en consideración las complicaciones neurológicas como el riesgo de hipertensión intracraneal, vasoespasmo e isquemia cerebral tardía y asegurar las medidas terapéuticas recomendadas para dichas complicaciones.35
Conclusiones
En los pacientes con STT, aunque la enfermedad previamente se había manejado como una entidad benigna, actualmente se debe reconocer como un compromiso orgánico con un impacto en la mortalidad a corto y mediano plazo, lo cual debe motivar la sospecha clínica en aquellos pacientes con HSAa.
La presente revisión bibliográfica examinó la evidencia actual en torno al STT en asociación con la HSA. Se han analizado los mecanismos fisiopatológicos, así como las manifestaciones clínicas y los estudios diagnósticos de mayor relevancia. Además, se han descrito las principales estrategias terapéuticas propuestas para abordar esta condición clínica.
Referencias
1. Chen Z, Venkat P, Seyfried D, Chopp M, Yan T, Chen J. Brain-heart interaction: cardiac complications After Stroke. Circ Res. 2017; 121:451-468. DOI: 10.1161/CIRCRESAHA.117.311170
2. Wagner S, Güthe T, Bhogal P, Cimpoca A, Ganslandt O, Bäzner H, Henkes H. Aneurysmal subarachnoid hemorrhage as a trigger for Takotsubo syndrome: a comprehensive review. Rev Cardiovasc Med. 2021; 22:1241-1251. DOI: 10.31083/j.rcm2204132
3. Baker C, Muse J, Taussky P. Takotsubo syndrome in neurologic nisease. World Neurosurg. 2021; 149:26-31. DOI: 10.1016/j.wneu.2021.01.139
4. Kaculini C, Sy C, Lacci JV, Jafari AA, Mirmoeeni S, Seifi A. The association of Takotsubo cardiomyopathy and aneurysmal subarachnoid hemorrhage: A U.S. nationwide analysis. Clin Neurol Neurosurg. 2022; 215:107211. DOI: 10.1016/j.clineuro.2022.107211
5. Elgendy AY, Elgendy IY, Mansoor H, Mahmoud AN. Clinical presentations and outcomes of Takotsubo syndrome in the setting of subarachnoid hemorrhage: A systematic review and meta-analysis. Eur Heart J Acute Cardiovasc Care. 2018; 7:236-245. DOI: 10.1177/2048872616679792
6. Murthy SB, Shah S, Rao CP, Bershad EM, Suarez JI. Neurogenic stunned myocardium following acute subarachnoid hemorrhage: pathophysiology and practical considerations. J Intensive Care Med. 2015; 30:318-325. DOI: 10.1177/0885066613511054
7. Cammann VL, Würdinger M, Ghadri JR, Templin C. Takotsubo syndrome: uncovering myths and misconceptions. Curr Atheroscler Rep. 2021; 23: 53. DOI: 10.1007/s11883-021-00946-z
8. Hasegawa Y, Uchikawa H, Kajiwara S, Morioka M. Central sympathetic nerve activation in subarachnoid hemorrhage. J Neurochem. 2022; 160:34-50. DOI: 10.1111/jnc.15511
9. Assad J, Femia G, Pender P, Badie T, Rajaratnam R. Takotsubo syndrome: a review of presentation, diagnosis and management. Clin Med Insights Cardiol. 2022; 16:11795468211065782. DOI: 10.1177/11795468211065782
10. Lyon AR, Citro R, Schneider B, Morel O, Ghadri JR, Templin C, Omerovic E. Pathophysiology of takotsubo syndrome: jacc state-of-the-art review. J Am Coll Cardiol. 2021; 77:902-921. DOI: 10.1016/j.jacc.2020.10.060
11. Dias A, Núñez Gil IJ, Santoro F, Madias JE, Pelliccia F, Brunetti ND, et al. Takotsubo syndrome: state-of-the-art review by an expert panel – Part 1. Cardiovasc Revasc Med. 2019; 20:70-79. DOI: 10.1016/j.carrev.2018.11.015
12. Ali A, Ahmad MQ, Malik MB, Alvi ZZ, Iftikhar W, Kumar D, et al. Neurogenic stunned myocardium: a literature review. Cureus. 2018 10;10: e3129. DOI: 10.7759/cureus.3129
13. Wybraniec M, Mizia-Stec K, Krzych L. Stress cardiomyopathy: yet another type of neurocardiogenic injury: ‘stress cardiomyopathy’. Cardiovasc Pathol. 2014; 23:113-20. DOI: 10.1016/j.carpath.2013.12.003
14. Schlossbauer SA, Ghadri JR, Scherff F, Templin C. The challenge of Takotsubo syndrome: heterogeneity of clinical features. Swiss Med Wkly. 2017; 147:w14490. DOI: 10.4414/smw.2017.14490
15. Yalta K, Yilmaztepe M, Zorkun C. left ventricular dysfunction in the setting of takotsubo cardiomyopathy: a review of clinical patterns and practical implications. Card Fail Rev. 2018; 4:14-20. DOI: 10.15420/cfr.2018:24:2
16. Fujita T, Nakaoka Y, Hayashi S, Imai RI, Nishida K, Seki SI, et al. incidence and clinical characteristics of Takotsubo syndrome in patients with subarachnoid hemorrhage. Int Heart J. 2022; 63:517-523. DOI: 10.1536/ihj.21-391
17. Pinnamaneni S, Dutta T, Melcer J, Aronow WS. Neurogenic stress cardiomyopathy associated with subarachnoid hemorrhage. Future Cardiol. 2015; 11:77-87. DOI: 10.2217/fca.14.73
18. Hwang EH, Koo JH, Lee YH, Song JH, Lim YC. Neurogenic pulmonary edema and Takotsubo cardiomyopathy in aneurysmal subarachnoid hemorrhage. Acta Neurochir (Wien). 2023; 165:3677-3684. DOI: 10.1007/s00701-023-05824-y
19. Abd TT, Hayek S, Cheng JW, Samuels OB, Wittstein IS, Lerakis S. Incidence and clinical characteristics of takotsubo cardiomyopathy post-aneurysmal subarachnoid hemorrhage. Int J Cardiol. 2014; 176:1362-1364. DOI: 10.1016/j.ijcard.2014.07.279
20. Medina de Chazal H, Del Buono MG, Keyser-Marcus L, Ma L, Moeller FG, Berrocal D, et al. Stress cardiomyopathy diagnosis and treatment: jacc state-of-the-art review. J Am Coll Cardiol. 2018; 72:1955-1971. DOI: 10.1016/j.jacc.2018.07.072
21. Singh T, Khan H, Gamble DT, Scally C, Newby DE, Dawson D. Takotsubo syndrome: pathophysiology, emerging concepts, and clinical implications. Circulation. 2022; 145:1002-1019. DOI: 10.1161/CIRCULATIONAHA.121.055854
22. Citro R, Okura H, Ghadri JR, Izumi C, Meimoun P, Izumo M, et al. Multimodality imaging in Takotsubo syndrome: a joint consensus document of the European Association of Cardiovascular Imaging (EACVI) and the Japanese Society of Echocardiography (JSE). Eur Heart J Cardiovasc Imaging. 2020; 21:1184-1207. DOI: 10.1093/ehjci/jeaa149
23. Santoro F, Mallardi A, Leopizzi A, Vitale E, Stiermaier T, Trambaiolo P, et al. Stepwise approach for diagnosis and management of Takotsubo syndrome with cardiac imaging tools. Heart Fail Rev. 2022; 27:545-558. DOI: 10.1007/s10741-021-10205-7
24. Cardin C, Roncalli J, Lairez O, Austruy J, Elbaz M, Carrie D, et al. Subarachnoid haemorrhage associated with midventricular Tako-Tsubo syndrome. Int J Cardiol. 2011; 146: e46-48. DOI: 10.1016/j.ijcard.2009.03.079
25. Shoukat S, Awad A, Nam DK, Hoskins MH, Samuels O, Higginson J, et al. Cardiomyopathy with inverted tako-tsubo pattern in the setting of subarachnoid hemorrhage: a series of four cases. Neurocrit Care. 2013; 18:257-260. DOI: 10.1007/s12028-011-9658-9
26. Thygesen K, Alpert JS, Jaffe AS, Chaitman BR, Bax JJ, Morrow DA, et al. Fourth universal definition of myocardial infarction (2018). J Am Coll Cardiol. 2018;72:2231-2264. DOI: 10.1016/j.jacc.2018.08.1038
27. Ghadri JR, Wittstein IS, Prasad A, Sharkey S, Dote K, Akashi YJ, et al. international expert consensus document on takotsubo syndrome (part i): clinical characteristics, diagnostic criteria, and pathophysiology. Eur Heart J. 2018; 39:2032-2046. DOI: 10.1093/eurheartj/ehy076
28. Ghadri JR, Wittstein IS, Prasad A, Sharkey S, Dote K, Akashi YJ, et al. international expert consensus document on takotsubo syndrome (part ii): diagnostic workup, outcome, and management. Eur Heart J. 2018; 39:2047-2062. DOI: 10.1093/eurheartj/ehy077
29. Zghyer F, Botheju WSP, Kiss JE, Michos ED, Corretti MC, Mukherjee M, Hays AG. Cardiovascular imaging in stress cardiomyopathy (Takotsubo syndrome). Front Cardiovasc Med. 2022; 8: 799031. DOI: 10.3389/fcvm.2021.799031
30. Citro R, Bellino M, Merli E, Di Vece D, Sherrid MV. Obstructive hypertrophic cardiomyopathy and Takotsubo syndrome: how to deal with left ventricular ballooning? J Am Heart Assoc. 2023; 12: e032028. DOI: 10.1161/JAHA.123.032028
31. Madias, John E. Insulin and short acting iv beta blockers: a “new” proposal for the acute management of Takotsubo syndrome. Int J Cardiol. 2021; 334: 18-20. DOI: 10.1016/j.ijcard.2021.04.033
32. Dias A, Núñez Gil IJ, Santoro F, Madias JE, Pelliccia F, Brunetti ND, et al. Takotsubo syndrome: state-of-the-art review by an expert panel – part 2. Cardiovasc Revasc Med. 2019; 20:153-166. DOI: 10.1016/j.carrev.2018.11.016
33. Molnár C, Gál J, Szántó D, Fülöp L, Szegedi A, Siró P, et al. Takotsubo cardiomyopathy in patients suffering from acute non-traumatic subarachnoid hemorrhage-A single center follow-up study. PloS One. 2022;17: e0268525. DOI: 10.1371/journal.pone.0268525
34. Zghyer F, Botheju WSP, Kiss JE, Michos ED, Corretti MC, Mukherjee M,et al. Cardiovascular Imaging in stress cardiomyopathy (Takotsubo syndrome). Front Cardiovasc Med. 2022; 8:799031. DOI: 10.3389/fcvm.2021.799031
35. Ran KR, Wang AC, Nair SK, Akça O, Xu R. Acute Multidisciplinary management of aneurysmal subarachnoid hemorrhage (aSAH). Balkan Med J. 2023; 40: 74-81. DOI: 10.4274/balkanmedj.galenos.2023.2023-1-100

Esta obra está bajo una licencia internacional: Creative Commons Atribución-NoComercial-CompartirIgual 4.0.